切除不能進行・再発大腸癌に対するRegorafenibの低用量開始/増量試験におけるプロスペクティブ評価
Prospective evaluation of regorafenib dose escalation strategy with low starting dose in patients with colorectal cancer.
Toshiki Masuishi, et al.
監修コメント
Regorafenib(REG)はその特徴的な有害事象の懸念から、本邦および米国より減量開始/増量計画の臨床試験が報告されてきた1,2)。
本試験は3つ目の減量開始試験の報告であるが、本試験ではREGを120mgで投与開始後、1サイクル目の15日目に増量基準に準じて160mgに増量する計画となっていたが、増量に至った症例はわずか9%であった。2018年1月のASCO-GIで報告された本邦からの増量試験1)では主治医判断で増量可能と規定されていたため、増量に至った症例割合はさらに低く1.7%であった。一方、米国での増量試験2)では多くの症例が増量に至っており、日本人と欧米人の毒性プロファイルの違いが本邦試験における増量割合の低さに影響している可能性が示唆される。
有効性に関して、本研究においては病勢制御割合(DCR)が32.4%と主要評価項目を達成することができなかった。また、安全性に関してはCORRECT試験の日本人サブセット3)と比較すると、手足の皮膚反応は若干低頻度であったものの、蛋白尿や高血圧、AST上昇に関してはむしろ高率に発現しており、減量開始戦略により有効性を担保し、有害事象のみを軽減するというコンセプトを実現し得るか少々疑問が残る結果であった。
一方、過去の2試験と大きく異なるポイントが血中濃度測定である。REGを増量できる症例(早期有害事象が生じない症例)では8日目のREG血中濃度が低いことが示された。今回の報告は増量できた症例における安全性と有効性であった。増量後の血中濃度測定の報告はなかったが、仮に、REGを増量することで血中濃度が上昇し有効性向上につながるのであれば、REGの血中濃度モニタリングが今後のREG投与に際してひとつの鍵になる可能性があると思われた。今後の報告に期待したい。
(コメント・監修:北海道大学病院 消化器内科 助教 結城 敏志)
本研究の特徴は、事前設定された投与量増量基準と薬物動態のモニタリング
Regorafenib(REG)の投与量を120mgから開始し、投与開始後15日目に160mgに増量し、その有効性と安全性を探索した試験結果が、舛石らによって発表された。本試験の特徴は、あらかじめ投与量の増量基準を設定したことと薬物動態をモニタリングしたことである。
対象は、組織学的に腺癌の診断がついた転移性結腸直腸癌で、フッ化ピリミジン系製剤、Oxaliplatin(OX)、Irinotecan(IRI)、Bevacizumab(Bmab)、Cetuximab(Cmab)、Panitumumab(Pmab、RAS野生型の場合)などによる標準治療中または投与終了後3ヵ月以内に病勢の増悪が認められた症例とし、ECOG PS(performance status)は0または1を適格とした。
本試験では、120mgでREGの投与を開始し、投与開始後15日目にあらかじめ設定された投与増量基準に従い160mgに増量された。この増量基準は、最初の14日間に投与中断や減量しなかった症例に適応された。また、REGとその活性代謝物であるM-2とM-5の血中薬物動態を観察するために、8日目、15日目、22日目に採血が行われた(図1)。
主要評価項目は病勢制御割合(DCR)とし、副次評価項目を、奏効割合(RR)、無増悪生存(PFS)期間、全生存(OS)期間、安全性として試験が行われた。統計学的解析は、DCRの閾値30%、期待値45%として、検出力80%、αエラー5%に設定し、必要症例数を67例と設定された。
この試験には68例が登録され、その患者背景は、年齢中央値64歳(範囲40~81歳)、性別は男性32例(47%)、女性36例(53%)、PSは0が37例(54%)、1が31例(46%)であった。原発部位は、盲腸-横行結腸14例(20%)、下行結腸-S状結腸20例(29%)、直腸34例(50%)であった。RAS遺伝子変異は、野生型と変異型がともに34例(50%)であった。前治療数は、1または2が26例(38%)、3が25例(37%)、4が17例(25%)であった。前治療で投与された薬剤の内訳は、フッ化ピリミジン系製剤100%、OX 96%、IRI 100%、抗VEGF抗体薬96%、抗EGFR抗体薬47%、FTD/TPI(TAS-102)40%であった。
図1 Study schema and treatment(発表者の許可を得て掲載)
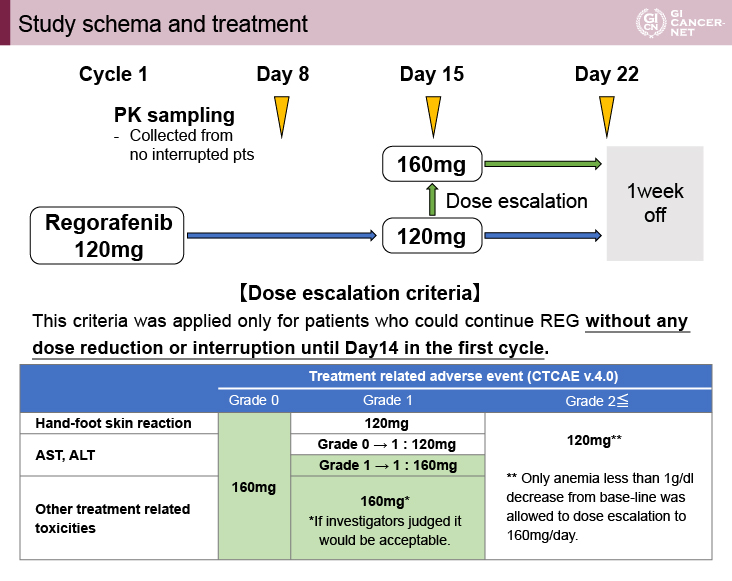
1サイクル目で増量は9%、減量は13%、投与中断は66%
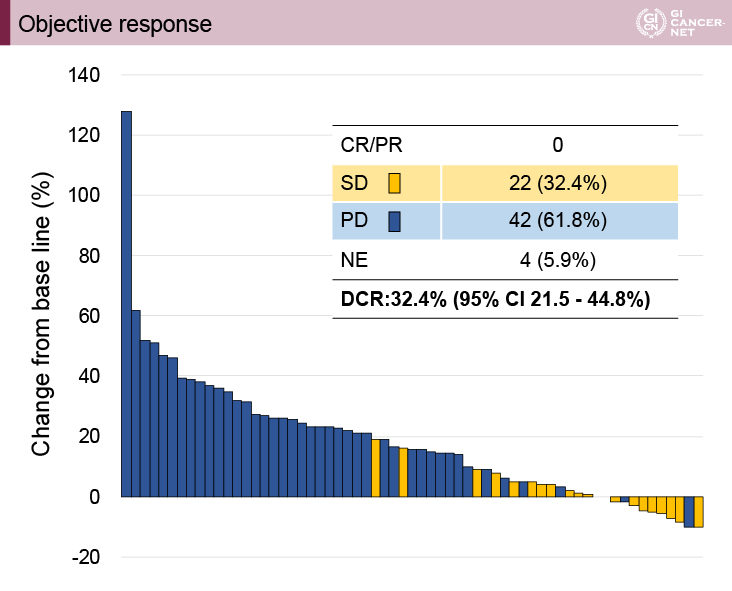
主要評価項目であるDCRは32.4%(95% CI: 21.5-44.8%)であった(図2)。PFS期間は1.8ヵ月(95% CI: 1.8-2.1)、OS期間は7.3ヵ月(95% CI: 6.0-11.5)であった。増量基準に従い1サイクル目で120mgから160mgに増量できた症例は6例(9%)であった。一方、1サイクル目で120mgから80mgに減量が必要であった症例は9例(13%)、投与中断となった症例は45例(66%)であった。
全サイクルにおけるREGに特徴的な有害事象は、手足の皮膚反応が全gradeで76%、grade 3以上で21%であった。AST/ALTの上昇は、全gradeで72%/49%、grade 3以上で12%/3%であった。また、蛋白尿/高血圧は全gradeで68%/69%、grade 3以上で18%/24%と報告された。
図2 Objective response(発表者の許可を得て掲載)
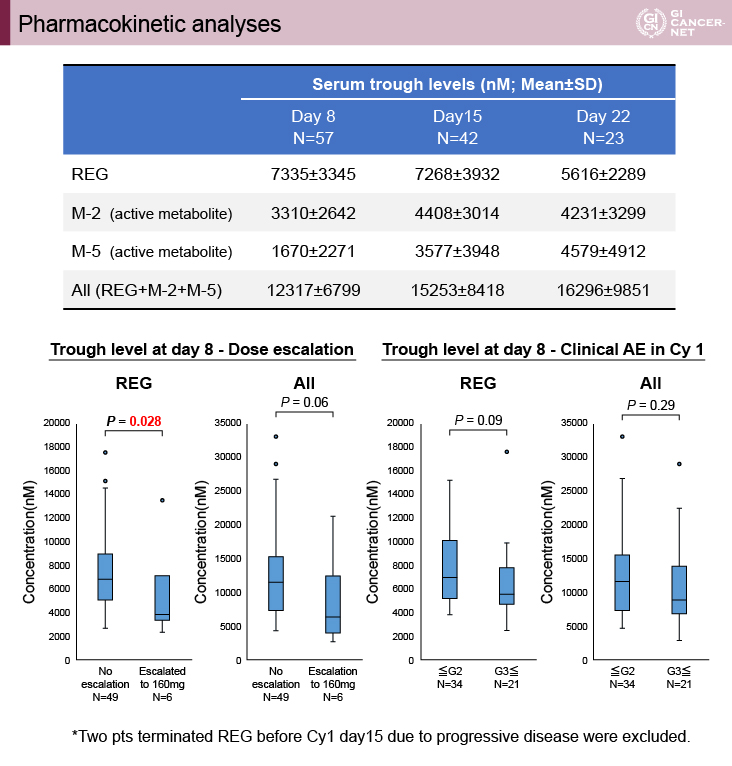
増量例では8日目のRegorafenib血中濃度が低かった
血中薬物動態の検討では、REGを増量しなかった症例と増量した症例で検討が行われ、増量例では8日目のREGの血中濃度が低かった(p=0.028)。代謝物を含めた解析では有意差を示さなかったが、血中濃度は低い傾向であった(p=0.06)。また、grade 2以下とgrade 3以上に分けた1サイクル目の有害事象の比較検討では、血中濃度に有意差は認めなかった(図3)。
図3 Pharmacokinetic analyses(発表者の許可を得て掲載)
(レポート:関西ろうさい病院 下部消化器外科 副部長 賀川 義規)
References
- 1) REGOC-12試験:Kudo T, et al.: ASCO-GI, 2018 #821[UMIN-CTR][ASCO Meeting Library]
- 2) ReDOS試験:Bekaii-Saab TS, et al.: ASCO-GI, 2018 #611[Journal of Clinical Oncology][ClinicalTrials.gov]
- 3) Yoshino T, et al.: Invest New Drugs. 33(3): 740-750, 2014[PubMed]
結城 敏志 先生
北海道大学病院 消化器内科 助教